Welcome to ChromOS
NGS Samples
Tissues (Cells)
Registered Users

ChromOS services online access to the data resources from Chromosome Orchestration System (OS) , a research project supported by MEXT Japan.
Chromosomes play a fundamental role in many biological processes. Previous research efforts have advanced our understanding of specific chromosomal events, such as DNA transcription, replication, recombination, partitioning, and epigenetic modification. One of the major future challenges in chromosome biology will be to provide an overall framework of how these individual activities are orchestrated and coordinated to maximize their effects in a variety of biological processes that evolve over time. The main goal of this project is to describe the mechanisms that regulate the functional unity of the chromosomes (chromosome OS) by thoroughly examining the structural relationship between, and the hierarchy of, individual chromosomal functions.
| Accession | Cell | Organism | Restriction enzyme | Condition | Run ID |
|---|---|---|---|---|---|
| OLP-2-620 | RPE | human | MboI | ESCO1, 2 KD (siRNA transfection 48 h), No Sync | 1809JNHX-0004_si11-19_res |
| OLP-2-614 | RPE | human | MboI | RAD21 KD (siRNA transfection 72 h), No Sync | 1809JNHX-0003_si621_res |
| OLP-2-628 | RPE | human | MboI | PDS5B KD (siRNA transfection 48 h), No Sync | HN00102383_si91_res |
| OLP-2-621 | RPE | human | MboI | MAU2 KD (siRNA transfection 72 h), No Sync | 1809JNHX-0004_si251_res |
| OLP-2-613 | RPE | human | MboI | RAD21 KD (siRNA transfection 72 h), No Sync | 1809JNHX-0003_si7-621_res |
| OLP-2-623 | RPE | human | MboI | CTCF KD (siRNA transfection 72 h), No Sync | 1811KHX-0110_R_626_628_d3_res |
| OLP-2-624 | RPE | human | MboI | WAPL KD (siRNA transfection 48 h), No Sync | HN00102380_si31_res |
| OLP-2-625 | RPE | human | MboI | PDS5A KD (siRNA transfection 72 h), No Sync | HN00102380_si88_d3_res |
| OLP-2-619 | RPE | human | MboI | ESCO1 KD (siRNA transfection 48 h), No Sync | 1809JNHX-0004_si11_res |
| OLP-2-626 | RPE | human | MboI | JQ1 (BRD Inhibitor), No Sync | HN00102383_JQ1_plus_res |
| OLP-2-615 | RPE | human | MboI | Control, No Sync | 1807JNHX-0018_Ctrl_res |
| OLP-2-627 | RPE | human | MboI | PDS5A,B KD (siRNA transfection 48 h), No Sync | HN00102383_si88_91_res |
| OLP-2-616 | RPE | human | MboI | Control, No Sync | 1809JNHX-0003_Ctrl_res |
| OLP-2-618 | RPE | human | MboI | CTCF KD (siRNA transfection 72 h), No Sync | 1811KHX-0109_FT_WT_res |
| OLP-2-617 | RPE | human | MboI | CTCF KD (siRNA transfection 72 h), No Sync | 1809JNHX-0003_si628_res |
| OLP-2-622 | RPE | human | MboI | Control, No Sync | 1811KHX-0109_R_Ctrl_res |
| OLP-2-629 | LCL | human | MboI | WT, No Sync | 1904JNHX-0009_GIA_res |
| OLP-2-630 | LCL | human | MboI | HP1beta mutation, No Sync | 1904JNHX-0009_RIM_res |
| OLP-2-631 | HCT116/Rad21-mAD/TIR1 | human | MboI | NIPBL, exon3 frame-shift, No Sync | 1903JNHX-0034_B3_res |
| OLP-2-633 | HCT116/ESCO1-mAD/TIR1 | human | MboI | Dox 12 h -> IAA 3 h (ESCO1 depletion), No Sync | 1907JNHX-0026_E1_dox_iaa_res |
| OLP-2-632 | HCT116/ESCO1-mAD/TIR1 | human | MboI | Dox 16 h -> (control), No Sync | 1908JNHX-0025_E1_dox_res |
| OLP-2-634 | HCT116 | human | MboI | NIPBL, exon 3 single allele mutation, No sync | 1807JNHX-0018_HCT_3-3_res |
| OLP-2-635 | HCT116 | human | MboI | Wild Type, No Sync | 1807JNHX-0018_HCT_Wt_res |
| OLP-2-641 | fibroblast | human | MboI | WT, female, No Sync | 1807JNHX-0018_GM2036_res |
| OLP-2-640 | fibroblast | human | MboI | WT, female, No Sync | 1904JNHX-0009_2036_res |
| OLP-2-639 | fibroblast | human | MboI | CdLS, NIPBL:2479_2480delAG; R827GfsX2, No Sync | 1807JNHX-0018_CdLS304_res |
| OLP-2-636 | fibroblast | human | MboI | CdLS, NIPBL: 1372C>T;Q458X / Nonsense, female, No Sync | 1807JNHX-0018_CdLS510_res |
| OLP-2-642 | fibroblast | human | MboI | WT, male, No Sync | 1807JNHX-0018_GM3348_res |
| OLP-2-638 | fibroblast | human | MboI | CdLS, NIPBL:2479_2480delAG; R827GfsX2, No Sync | 1807JNHX-0018_CdLS087_res |
| OLP-2-637 | fibroblast | human | MboI | WT, male, No Sync | 1807JNHX-0018_CdLS006_res |
| OLP-2-643 | Blood Cell Stem Cell | mouse | MboI | STAG2 conditional KO | 1811KHX-0110_KO_561_res |
| OLP-2-646 | Blood Cell Stem Cell | mouse | MboI | STAG2 conditional KO | 1811KHX-0110_KO_544_res |
| OLP-2-645 | Blood Cell Stem Cell | mouse | MboI | WT | 1811KHX-0110_WT_546_res |
| OLP-2-644 | Blood Cell Stem Cell | mouse | MboI | WT | 1811KHX-0110_WT_563_res |
| OLP-2-654 | 293FT | human | MboI | WT, No Sync | 1905JNHX-0006_FT_res |
| OLP-2-648 | 293FT | human | MboI | lentiviral vector Infection, Control, No Sync | 1903JNHX-0034_FT-vi24_res |
| OLP-2-649 | 293FT | human | MboI | lentiviral vector Infection, Control, No Sync | 1903JNHX-0035_FT-Wt_res |
| OLP-2-651 | 293FT | human | MboI | lentiviral vector Infection, Control, No Sync | 1908JNHX-0026_vi24_res |
| OLP-2-652 | 293FT | human | MboI | Wild AFF4 lentiviral vector Infection, No Sync | 1908JNHX-0026_vi27_res |
| OLP-2-647 | 293FT | human | MboI | Mutant AFF4 lentiviral vector Infection, No Sync | 1903JNHX-0035_FT-vi28_res |
| OLP-2-653 | 293FT | human | MboI | Mutant AFF4 lentiviral vector Infection, No Sync | 1908JNHX-0026_vi28_res |
| OLP-2-650 | 293FT | human | MboI | NIPBL, exon 3 bi-allele mutation, No Sync | 1811KHX-0109_FT_ND1_res |
- ChromOS manages the data from the project "Chromosome OS".
- ChromOS provides the analytic pipeline for Hi-C data by collaborating with OpenLooper (OLP).
- Registration via OpenLooper is required. Thereby, users can manage the data and submit jobs to analyze. Learn more..
- Anyone can freely access the data opened by the registered users.
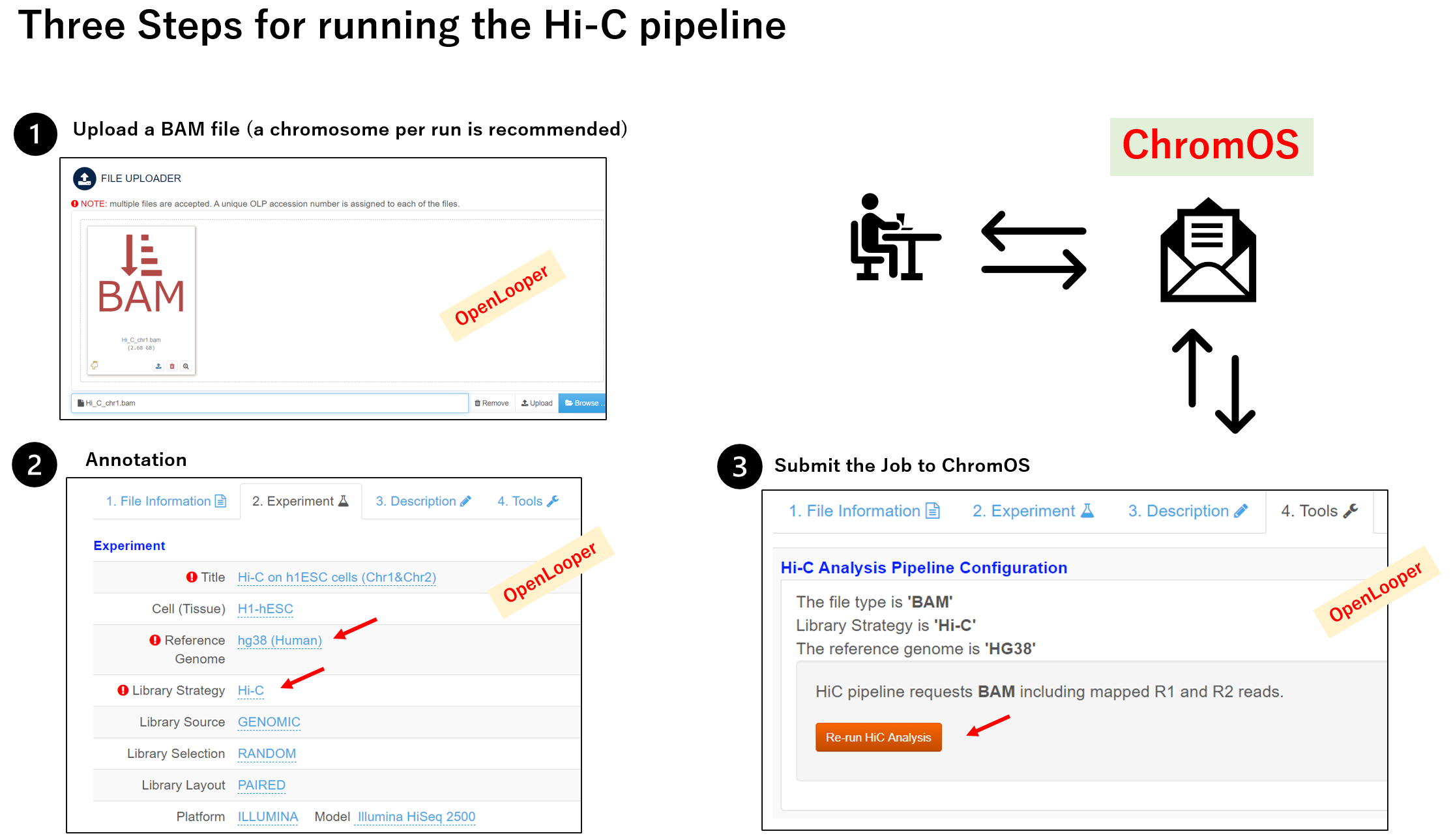
To reduce BAM filesize, prepare one chromosome per BAM.
"Samtools view" command will be helpful to do it.
Data Sharing (OpenLooper)
OpenLooper (OLP) collects genome-wide data on chromatin structures investigated by various high-throughput experimental assays. Simultaneously, OLP provides a platform that supports opening and sharing the data.
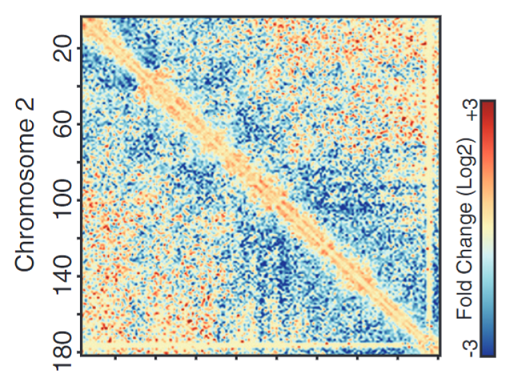
Hi-C Analysis
ChromOS runs a pipeline for Hi-C data analysis with user-uploaded BAM files via OpenLooper.
This pipeline is now in service (2020.06).
Example: [OLP-1-608]
HiC collection: [RPE, HCT116, LCL,...]
![]()



